In a counterintuitive twist that challenges established views on cellular biology, researchers have discovered that suppressing a gene typically considered essential for life may hold the key to treating a debilitating neurodegenerative disease. A groundbreaking study published just two days ago in the journal Nature indicates that reducing levels of the mitochondrial protein Ferredoxin-2 (FDX2) significantly aids cell survival in models of Friedreich's ataxia.
The findings present a complex medical paradox. While FDX2 is known to be critical for preventing tumors and maintaining lipid homeostasis, its reduction appears to suppress the lethal effects of frataxin deficiency, the root cause of Friedreich's ataxia. This discovery opens a new, albeit risky, frontier for therapeutic intervention, suggesting that cellular health is less about the absolute presence of a protein and more about the delicate balance of mitochondrial machinery.
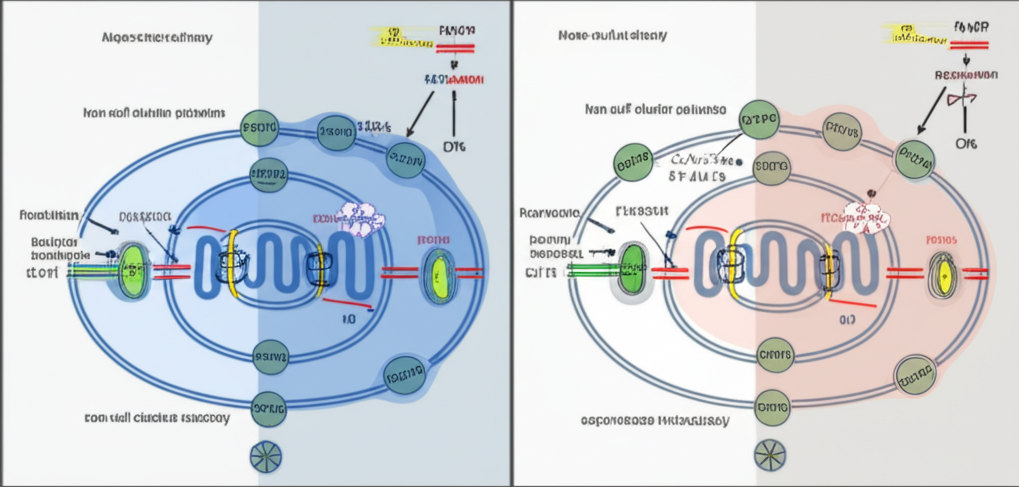
The Mechanism of Rescue
Friedreich's ataxia is a rare genetic disease that causes progressive damage to the nervous system and movement problems. It stems from a deficiency in frataxin, a mitochondrial protein involved in the assembly of iron-sulfur clusters-tiny cofactors essential for energy production and DNA repair.
According to the new data released in Nature, scientists utilized a forward genetic screen in Caenorhabditis elegans (roundworms) to identify genetic modifiers of this condition. They found that decreased levels of FDX2 suppressed the loss of frataxin in both worm and mouse models. The mechanism involves the relief of FDX2's inhibition of NFS1 activity. Essentially, in a healthy system, FDX2 and frataxin work in concert; however, when frataxin is missing, FDX2 becomes an impediment. Removing or reducing it allows the remaining machinery to function more effectively, restoring viability to the organism.
"Decreased levels of the ferredoxin FDX2 suppress the loss of frataxin in worms and in mice by relief of FDX2 inhibition of frataxin-stimulated NFS1 activity." - Nature, 2025
The Double-Edged Sword of FDX2
While the potential to treat Friedreich's ataxia is promising, the manipulation of FDX2 is fraught with danger. Background research clarifies that FDX2 is not merely a bystander gene. A study published in ScienceDirect in late 2024 highlighted that FDX2 is "indispensable" for tumor suppression and lipid homeostasis. In ovarian cancer cells, the loss of FDX2 induced iron overload and DNA damage, driving a program of cellular senescence (aging) and death.
This creates a significant challenge for drug developers. The therapeutic window is narrow: reduce FDX2 enough to alleviate ataxia symptoms, but not so much that it triggers tumor growth or ferroptosis (a type of iron-dependent cell death). Experts indicate that FDX2 acts as an electron transport protein required for biosynthesis, meaning its total removal is catastrophic for a healthy cell, even if its partial reduction helps a diseased one.
Clinical and Commercial Implications
The business and medical implications of this discovery are immediate for the rare disease sector. Therapies targeting mitochondrial disorders are notoriously difficult to develop due to the organelle's complex biochemistry. This finding suggests a new class of "inhibitor therapies" could be viable for metabolic diseases previously thought untreatable.
- Target Identification: Biotech firms focusing on Friedreich's ataxia may pivot to screen for FDX2 inhibitors rather than solely focusing on frataxin replacement.
- Safety Protocols: Any resulting therapy will require rigorous safety monitoring for oncogenic side effects, given the tumor-suppression role of FDX2.
- Broader Applications: Understanding FDX2's role in ferroptosis could lead to insights into cancer treatments where inducing cell death is the goal, rather than the risk.
Future Research Directions
Looking ahead, the path from the laboratory to the clinic will involve navigating conflicting biological signals. Recent papers from PubMed and Nature Chemical Biology emphasize that while FDX2 and its counterpart FDX1 have distinct roles, there is some functional overlap in electron donation. Future research will likely focus on dissociating the survival-promoting effects of FDX2 reduction from its cancer-preventing duties.
Furthermore, the connection between FDX2 mutations and mitochondrial myopathy underscores the need for precision medicine. Patients with natural variations in FDX2 show distinct severity in iron regulation, suggesting that any therapy must be highly personalized.
This discovery represents a quintessential moment in modern genetics: the realization that the cure for one ailment may lie in the suppression of a mechanism critical for another, forcing science to walk a tightrope toward recovery.






